




서 론
사시나무(Populus davidiana Dode)는 우리나라에서 자생하고 있는 사시나무속 가운데 그 분포 면적이 가장 넓어 사시나무절(Leuce)을 대표하는 수종이며, 근계형성 능력이 뛰어나 산 정상을 제외한 여러 환경에서 생존력이 강한 특성이 있다(Koo and Yeo, 2003). 사시나무의 유전체 개수는 총 19쌍으로(Kim et al., 2020), 유전체 크기는 옥수수의 1/6, 테다소나무의 1/40 수준인 520 mbp 정도로 비교적 그 크기가 작아서 유전체 연구가 활발히 진행되고 있다(Tuskan et al., 2004). 사시나무 유전체에 대한 물리지도(Kelleher et al., 2007) 및 엽록체 DNA 염기서열(Choi et al., 2017), 전장 유전체 해독을 통한 유전적 분화(Hou et al.,2020) 등 다양한 유전정보에 대한 자료가 축적되고 있어 임목 모델종으로서 인정받고 있으며 분자 유전학적으로도 중요한 수종이다(Bradshaw et al,. 2000).
유전연관지도(genetic linkage map)는 서로 다른 표현형을 가진 개체의 차대를 이용하여 차대에서 보이는 형질을 조사하여 부모 개체에서 가지고 있는 유전자의 상대적인 위치를 나타낸 지도이다. 차대에서 염색체 교차 및 재조합이 많이 일어난 것은 유전자 간의 거리가 멀다고 판단하고, 염색체 교차 및 재조합이 적게 나타난 것은 유전자 간의 거리가 가깝다고 판단하여 유전자 교차 및 재조합률을 계산하여 유전자의 상대적인 위치를 나타낸다(Liu, 2017). 이렇게 형질에 대한 유전자의 상대적 위치 정보를 가지고 있는 유전연관지도는 다양한 종류의 DNA 마커가 개발되기 시작하면서 망고, 밀의 육종교배(Kuhn et al., 2017; Devo et al., 1993)를 포함한 광범위한 육종사업에 이용되고 있다(Doergem, 2002; Maloofm, 2003).
차세대 염기서열 분석기술(next generation sequencing, NGS) 방법이 보편화되면서 기존에 사용하던 AFLP, RAPD 마커 등을 이용한 유전체 분석 방법보다 SNP (single nucleotide polymorphism) 마커를 이용한 분석기술은 임목에서도 고밀도 유전연관지도 작성이 가능하게 되었다(Davey et al., 2011). 이를 이용하여 사시나무속 수종에서는 기존 저밀도 형태로 작성되었던 유럽사시나무(P. tremula) 그리고 미루나무(P. deltoides)와 당버들(P. simonii)을 인공교배한 육종집단에서 고밀도 유전연관지도가 재작성되었다(Mousavi et al., 2016; Tong et al., 2016). 따라서 사시나무 역시 우리나라를 대표하는 사시나무속 수종으로서 고밀도 유전연관지도 작성이 다시 이루어져야 할 필요성이 대두되었다.
임목은 한 세대가 길고 유전검정을 실시하기 위해 많은 개체 수와 넓은 대규모의 시험지가 필요하게 된다. 반면에 적은 개체로도 목표 형질의 유전형을 밝혀내고 우수한 개체를 조기에 선발할 수 있는 유전연관지도 작성 및 양적형질 유전자좌(quantitative trait loci, QTLs) 탐색의 중요성이 주목받고 있다(Sewel and Neale, 2000). 아울러 최근 기후변화로 인하여 임목의 서식지, 생장 특성이 변화하고 있고 기온 상승으로 인한 식엽성 해충의 피해가 늘어나면서(Pureswaran et al,, 2018) 이를 대비하기 위한 형질 개량도 필요하다.
본 연구에서는 모수를 오대19로 하고 화분수를 봉현4로 하여 얻은 사시나무 4년생 F1 육종집단을 연구재료로 선정하고, 선정한 육종집단의 유전자형을 확인한 후 차세대 염기서열 분석 방법 가운데 하나인 Genotyping-by-sequencing (GBS) 기법을 사용하여 사시나무의 고밀도 유전연관지도 작성을 목적으로 하였다. 또한 사시나무 육종집단의 수고, 근원경 생장 그리고 식엽성 해충피해 회복력 형질에 관여하고 있을 것으로 추정되는 양적형질 유전자좌(QTLs)를 탐색하고, 전장유전체연관연구(genome-wide association study, GWAS)를 통해 조사한 형질에 대응하는 유전자를 탐색하고자 하였다.
재료 및 방법
경기도 수원시 권선구에 위치한 서울대학교 칠보산 학술림의 비가림하우스에 조성된 사시나무 육종집단을 실험 대상으로 하였다. 사시나무 육종집단은 표준유전체 작성 대상인 오대19호를 모수로 하고 봉현4호를 화분수로 하여 생산된 인공교배 F1 차대 집단이다(Figure 1). 사시나무 유전체 육종집단은 2018년도 인공 교잡하여 얻은 종자를 파종하여 2019년도에 한 개체 당 Polypropylene 재질의 내경 230 mm, 높이 245 mm의 포트에 이식되어 생장하고 있는 4년생 사시나무 298개체로 구성되어 있다.

사시나무 육종집단 298본 중 고사하지 않은 295본의 개체를 대상으로 2020년 3월 말에서 4월 초 새로 돋아나는 잎을 대상으로 엽 시료를 채취하였으며, 같은 시기에 고사한 3개체를 포함한 298본 전체 육종집단을 대상으로 수고 및 근원경 생장량을 측정하였다. 수고는 줄자를 사용하여 cm 단위로 측정하였고, 근원경은 디지매틱 캘리퍼스(Misutoyo, 500-182-30)를 사용하여 mm 단위의 소수점 둘째 자리까지 측정하였다.
2020년 7월 중순 사시나무 육종집단 내 모든 개체에 대하여 꼬마버들재주나방(Clostera anachoreta) 유충으로 인한 해충피해가 있었다(Figure 2), 해충 구제 농약인 스미치온을 20 L당 20 ml을 혼합하여 분무기를 이용해 충분히살포한 후 일주일이 지난 시점에서 각각의 개체가 꼬마버들재주나방 피해에서 회복하는 정도를 0~3점으로 나누어 점수화하였다. 0점은 고사한 개체, 1점은 피해를 받은 다음 회복이 불량한 개체, 2점은 피해를 받은 후 회복상태가 보통인 개체, 3점은 피해를 받았지만 회복상태가 우수한 개체로 구분하여 조사하였다.

수고 생장, 근원경 생장 및 꼬마버들재주나방 해충피해 회복력 등 세 가지 형질 간의 상호 연관성을 파악하기 위해 R software version 4.1.1 (R Project for Statistical Computing)을 이용하여 상관분석을 수행하였다.
선정된 사시나무 육종집단이 실제로 모수를 오대19로 하고 화분수를 봉현4로 한 인공교배집단이 맞는지 확인 및 검증하기 위하여 microsatellite 마커를 활용하여 유전자형을 확인하였다.
유전자형 확인을 위해 우선 DNA추출을 실행하였다. 사시나무 육종집단 295개체, 모수인 오대19 그리고 화분수인 봉현4에서 채취한 잎(30 mg~50 mg)을 지름 2 mm, 4 mm의 스테인리스 비드 2개와 함께 원심분리기용 소형 튜브에 넣고 액체질소에 약 30초간 급속 냉동한 후 TissueLyser II (Quiagen)를 이용하여 3분간 분쇄하였다. 분쇄되어 분말이 된 잎은 QIAGEN DNeasy Plant Mini Kit의 프로토콜에 따라 DNA를 추출하였다. 추출한 DNA는 NanoDrop™2000 (Thermo Scientific)을 사용해 DNA의 농도를 측정하였다.
추출된 DNA는 microsatellite 마커를 통해 유전자 증폭(PCR)을 수행하여 유전자형을 확인하였다. 유전자형을 확인하기 위해서는 최소 5개의 마커를 사용하는 것이 바람직하므로(Hardy et al., 2003), 이를 위하여 사시나무 근연종의 microsatellite 마커 중 선행연구(Lee et al., 2011)에서 사용된 ORPM26, ORPM312 (Tuskan et al., 2004), PMGC2501 (Cole, 2005), WPMS15(Smulders et al., 2001) 및 PTR4 (Dayanandan et al., 1998) 등 총 5개의 마커를 선정하여 사용하였다(Table 1).
모든 혼합물은 95°C에서 15분간 가열한 후 TaKaRa PCR Thermal Cycler Dice® TP600를 이용해 PCR을 진행하였다. ORPM26와 ORPM312 마커가 포함된 DNA 혼합물은 94°C에서 30초, 60°C에서 30초, 72°C에서 30초 과정을 10번 반복하였다. PMGC2501 마커가 포함된 DNA 혼합물은 94°C에서 30초, 52°C에서 30초, 72°C에서 1분 과정을 10번 반복하였으며, PTR4 및 WPMS15 마커가 포함된 DNA 혼합물은 94°C에서 30초, 57°C에서 30초, 72°C에서 1분 과정을 10번 반복하였다. 이후 모든 혼합물에 대하여 M-13 마커를 결합시키기 위해 94°C에서 30초, 52°C에서 30초, 72°C에서 1분간 진행하는 과정을 30번 반복하였다.
위와 같은 단계로 얻어진 PCR 증폭산물은 GeneScan™-500 ROX™ size standard를 포함한 Hi-Di Formamide를 첨가하였다. 만들어진 혼합물은 ABI 3730xl을 이용해 전기영동을 실시한 후 증폭산물 내 형광물질을 탐지하여 유전자형을 분석하였다. Geneious Prime®2021.1.1. (Biomatters) 프로그램을 통해 증폭산물의 크기를 기준으로 그 양을 나타낸 피크(peak)를 확인하였다. 이때, 모수 및 화분수와 같지 않은 피크를 가지고 있는 개체는 친자가 아닌 것으로 간주하여 연구대상에서 제외하였다.
추출한 DNA는 Quant-iT PicoGreen dsDNA Assay Kit (Molecular Probes, Eugene, OR, USA)의 표준 절차에 따라 정량화하였고, 이 과정에 Synergy HTX Multi-Mode Reader (Biotek, Winooski, VT, USA)를 이용하였다. 이후 제한효소를 사용한 DNA 절단, 어댑터 부착, 라이브러리 제작, 그리고 DNA 염기서열화(sequencing) 과정이 수행되었다. GBS를 이용한 DNA 라이브러리 제작은 De Donato et al.(2013)과 Elshire et al.(2011)에서 제시된 방법을 따랐다(Table 2).
절단된 DNA 절편들은 PRI select Beeds (Beckman Coulter. Brea, California, United States)를 사용하여 400 bp에서 700 bp (pick size: 약 450 bp) 사이에서 크기를 선별하였고, PCR을 통해 이들의 절편을 생성하였다. 생성된 GBS DNA 라이브러리는 Agilent 4200 TapeStation (Agilent Technologies, USA)에서 DNA 절편 크기 분포를 평가하였다. GBS 라이브러리는 Illumina NextSeq500 (Illumina, USA)를 이용하여 151 bp 길이의 Paired-end DNA 염기서열 분석이 완료되었다.
DNA 염기서열 분석 과정이 완료된 데이터의 전처리는 먼저 Stacks, FastQC, Cutadapt software (Andrews, 2010; Martin, 2011; Catchen et al., 2013)를 사용하여 수행하였다. Demultiflexing은 Stacks v2.55 내의 ‘process_radtags’ 모듈을 사용하여 ApeK I 제한효소에 대한 바코드 염기서열에 따라 각 샘플별로 수행하였다. 그 후, FastQC 소프트웨어를 사용하여 분석한 출력데이터의 기본 단위 보정 및 잠재적 어댑터 염기서열 제거를 위한 품질 검사를 수행하였고, 품질검사가 시행된 데이터들은 Trimmomatic v0.39 소프트웨어를 이용하여 어댑터 염기서열로 추정되는 부분을 제거하였다. 그런 다음 BWA v0.7.17 소프트웨어를 사용하여 출력데이터값을 Populus trichocarpa 참조유전체(P. trichocarpa v4.1 Nisqually-1)에 매핑하였다(Tuskan et al., 2006; Langmead and Salzberg, 2012).
사시나무 오대19×봉현4 F1 육종집단을 대상으로 PLINK v1.9의 ‘-pca’옵션을 사용하여 주성분 분석(principal components analysis)을 진행하였고, Admixture 1.3.0 소프트웨어를 이용하여 최대 가능도(maximum likelihood)를 각 개체에 대하여 추정하였다.
확보된 SNP 마커를 이용하여 Lep-MAP3 (Rastas, 2017) 프로그램을 이용하여 유전연관지도 구축을 진행하였으며, R/qtl 패키지로 작성된 유전연관지도를 이용하여 composite interval mapping을 수행하였다. 추출된 SNP 마커를 experimental crosses에서의 양적형질 유전자좌에 정렬하였다. 이후 선형 회귀분석으로 수고, 근원경 및 해충피해 회복력에 관련된 표현형과 유전형 간의 연관성 검정을 수행하였고, 1000번의 순열 테스트로 유의수준(α=0.05)의 LOD (logarithm-of-odds) score threshold 값을 추정하여 양적형질 유전자를 탐색하였다.
필터링으로 추출된 SNP 마커를 이용하여 수고, 근원경 및 해충피해 회복력의 표현형에 따른 GWAS를 수행해 양적형질 유전자좌에 정렬된 SNP의 위치를 재확인하였다. 여기에는 샘플들의 유전형과 표현형 데이터의 연관성을 보는 일반선형모델(general linear model) 방법, 샘플들 간의 관계까지 고려한 혼합선형모델(mixed linear model) 방법을 이용하였다. Tassel 소프트웨어와 Admixture 소프트웨어를 사용하여 얻은 kinship과 structure의 정보를 추가로 이용해 GWAS를 수행하였고, 가장 적합한 모델을 선정한 후 Bonferroni correction을 수행하여 모델에 연관분석으로 나온 유의미한 SNP 마커를 추출하였다.
결과 및 고찰
사시나무 육종집단에서 가장 수고가 높은 개체의 크기는 170 cm이었고 가장 작은 개체는 26 cm였으며, 총 298본의 수고 평균은 101.7 cm이었다. 근원경이 가장 굵은 것은 12.94 mm, 가장 가는 것은 2.87 mm였으며, 근원경 평균은 7.64 mm였다(Figure 3). 꼬마버들재주나방 피해 회복력에 있어서 0점인 개체가 33개체, 1점인 개체가 77개체, 2점인 개체가 99개체 그리고 3점인 개체가 89개체였다. 꼬마버들재주나방 피해에 대한 해충피해 회복 능력 형질의 평균은 1.82점이었다.
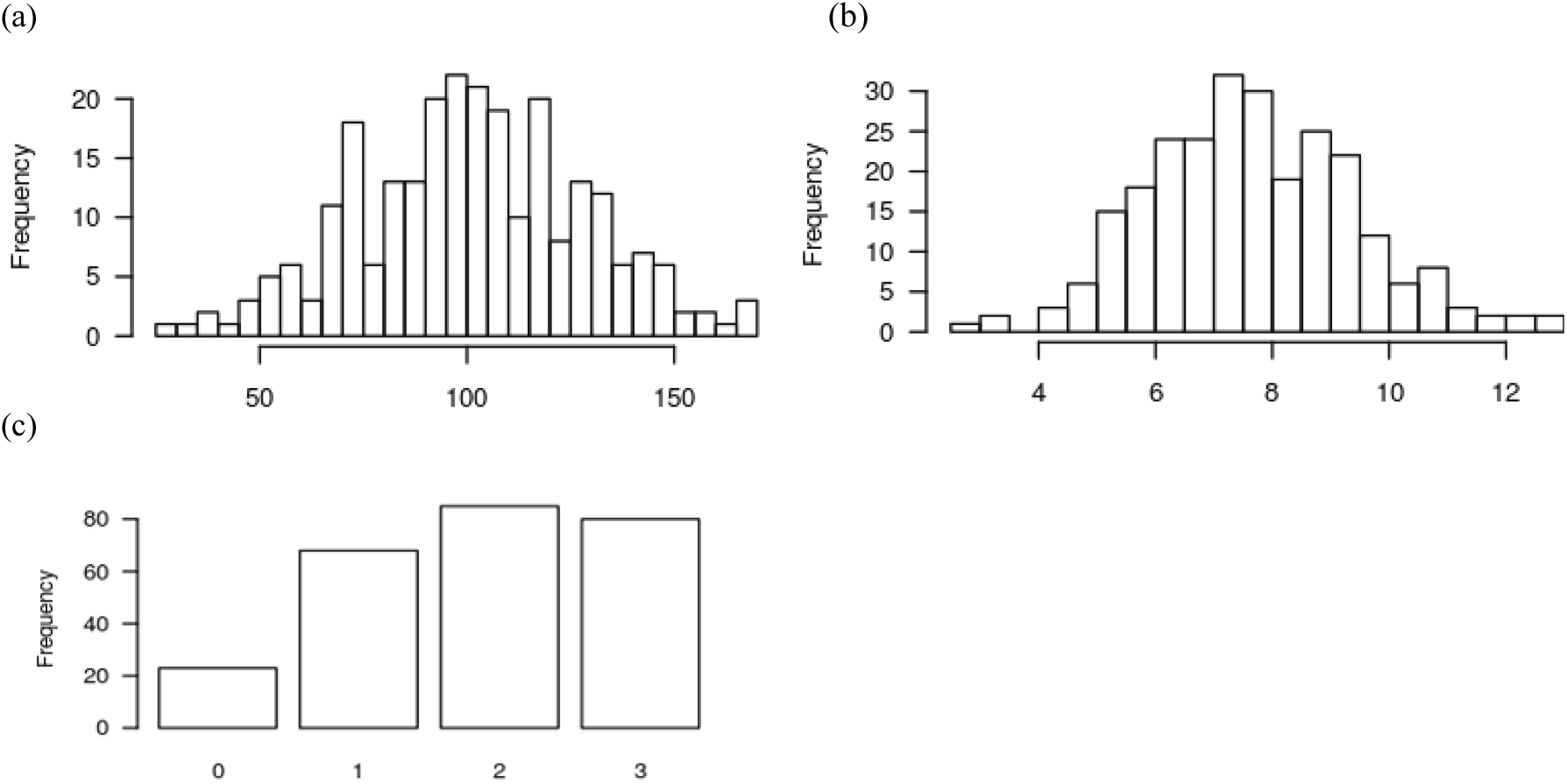
수고 및 근원경 생장 형질에 대하여 그 빈도를 그림 3과 같이 막대그래프 형태로 나타내었고, 조사 값이 다양하지 않은 꼬마버들재주나방 피해 회복력에 대한 형질을 제외하고 모두 정규분포를 따르고 있는 것을 확인할 수 있었다.
수고와 근원경 생장 그리고 해충피해 회복력 형질 간의 상관관계를 분석하였을 때 수고와 근원경 생장은 서로 높은 정의 상관관계(r=0.764)를 보였다. 그러나 꼬마버들재주나방 피해 회복력은 수고와 근원경 생장량과 서로 상관(−0.000, 0.013)을 나타내지 않았다.
Microsatellite 마커를 이용하여 유전자형을 확인한 결과, 모수인 오대19와 화분수인 봉현4는 ORPM26, PTR4, WPMS15 프라이머에서 각각 2개의 대립유전자를 가지고, ORPM312와 PMGC2501 프라이머에서 각각 3개의 대립유전자를 가지고 있었다. 연구에 사용된 오대19와 봉현4의 F1육종집단은 ORPM312, PTR4 프라이머에서 육종집단의 모든 개체의 유전자형이 오대19, 봉현4와 동일하게 나타났으나, ORPM26 프라이머에서 1개체, PMGC2501 프라이머에서 2개체 그리고 WPMS15 프라이머에서 28개체가 모수인 오대19, 화분수인 봉현4에서 관찰되지 않은 유전자형을 나타내었다. 따라서 유전자형 확인에서 탈락한 개체들을 친자가 아닌 것으로 간주해 연구대상에서 제외하여 총 264개체를 연구대상으로 선정하였으며, 이후 DNA 품질 검사에서 탈락한 4개체를 제외하여 총 260개체를 연구시료로 선정하였다.
부모 개체(오대19, 봉현4)와 차대를 포함한 총 262개체의 사시나무에 대해서 Populus trichocarpa 참조유전체 정렬을 통한 변이 추출을 진행하였을 때, 모수와 화분수인 오대19와 봉현4에서 총 1,717,613개의 원시 변이체(raw variant)가 발견되었다. 이후 원시 변이체는 참조유전체 조건에 따라 필터링하였고 참조유전체에 매핑되지 않은 73개의 scaffold를 포함하여 총 58,040개의 SNP 마커를 확보하였다(Table 3).
확보된 58,040개의 SNP 마커 중 유전연관지도 작성을 위해 부모 개체의 유전형과 비교하였을 때 손실이 존재하거나 monomorphic homozygous 및 segregation distortion을 보이는 SNP 마커를 제거하여 최종적으로 17,755개의 SNP 마커만 추출한 후 유전연관지도 작성에 사용하였다. Kosambi map function을 이용해 추출한 마커 간 거리를 계산하였고, LOD score가 5일 때 19개 연관군으로 나누어지는 유전연관지도를 얻을 수 있었다(Figure 4).
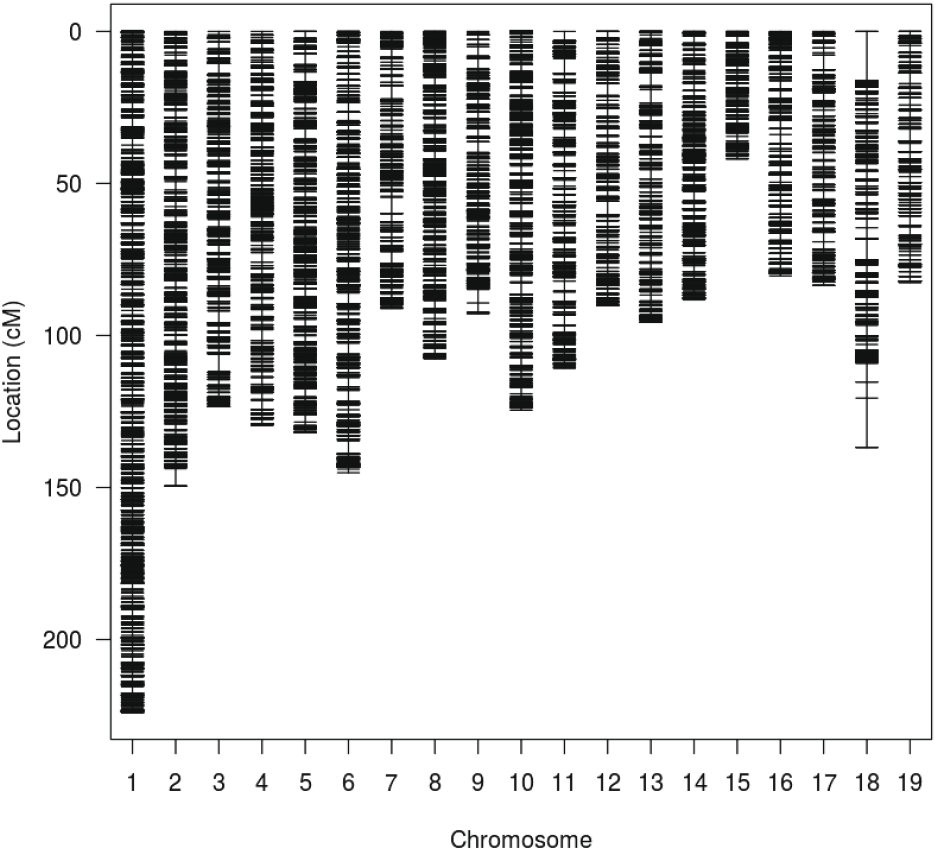
작성된 유전연관지도의 전체 길이는 2,129.54 cM이었으며, 가장 긴 연관군은 1번 연관군(LG1)으로 223.88 cM이었고 길이가 가장 짧은 연관군은 15번 연관군(LG15)으로써 41.91 cM이었다. 그리고 마커의 평균 밀도는 0.120 cM으로 나타났다(Table 4).
사시나무 육종집단을 대상으로 AFLP (amplified fragment length polymorphism) 마커를 이용하여 저밀도의 유전연관지도가 작성된 바 있으나(Kim et al., 2004), 본 연구에서는 microsatellite 마커를 이용하여 사시나무 F1 육종집단(모수 오대19 × 화분수 봉현4)의 친자 개체만을 사용하여 유전연관지도를 작성하였다. 즉, 조성된 육종집단 사시나무 295개체 중 31개체가 친자가 아닌 것으로 판단되어 해당 개체를 연구대상에서 제외하였다. 기존의 유전연관지도 연구(Kim et al., 2004; Tong et al., 2016; Zhigunov et al., 2017)에서는 친자 확인을 하지 않았다. 또한 본 연구에서 유전연관지도 작성에 사용된 개체의 수가 부모 개체를 포함하여 262개체로, 과거 유전연관지도 작성에 사용하는 집단의 크기가 100~200개체인 것을 고려하였을 때 그 집단의 크기가 약 0.3배에서 1.6배 더 크고 정확도가 더 높은 유전연관지도가 작성되었다.
본 연구의 사시나무 인공교배 육종집단에서 작성된 유전연관지도는 19개의 연관군(염색체)으로 나눠진 것을 확인할 수 있었는데, 이것은 FISH (fluorescent in situ hybridization)로 확인된 사시나무 핵형의 염색체 개수인 19쌍과 같은 결과이다(Kim et al., 2020). 기존에 작성된 사시나무속 수종의 미루나무(4,057 cM), 당버들(3,809 cM), 미국사시나무(3,054.99 cM) 등의 유전연관지도 길이(Tong et al., 2016; Zhigunov et al., 2017)와 비교할 때 우리나라 사시나무의 유전연관지도의 크기는 그보다 작은 2,129.54 cM로 나타났다.
사시나무 유전연관지도 작성에 사용된 SNP 마커의 개수는 총 17,755개이었는데, 이는 기존에 연구되었던 당버들, 미루나무 및 유럽사시나무의 유전연관지도 작성에 사용된 마커 수가 1,430개, 2,012개 및 2,055개인 것과 비교하면 약 7.6배에서 11.3배 더 많은 숫자이다. 따라서 마커 밀도로 표현되는 마커 간 평균 거리 역시 기존의 유전연관지도의 경우, 평균 2.65 cM에서 4.06 cM의 밀도를 가지고 있었으나, 본 연구에서 작성된 유전연관지도의 마커 밀도는 평균 0.12 cM로써 매우 고밀도의 유전연관지도가 작성되었다.
양적형질 유전자좌를 탐색하기 위해 수고, 근원경 및 해충피해 회복력 형질에 대한 1,000번의 순열검정(permutation test)으로 유의수준 α=0.05의 LOD 한계치(threshold score)를 추정하였다. 각 형질에 대해서 추정된 LOD 한계치는 각각 5.0, 5.1 및 5.1이었지만, 수고와 근원경 형질에 있어서 LOD score threshold를 넘는 양적형질 유전자좌는 관측되지 않았다.
본 연구에서 수고 및 근원경 생장 형질에 대한 양적형질 유전자좌(QTLs)가 확인되지 못한 이유로는, 육종집단의 수령이 아직 어려서 생장과 관련된 유전자의 발현이 뚜렷하지 않았을 수 있고(Day et al., 2001; Woo et al., 1994), 연구에서와 같이 식엽성 해충의 피해에 따른 생장 관련 유전자의 발현 및 탐색이 어려웠을 수 있다. 특히, 형질의 발현은 유전형 뿐만 아니라 육성과정의 환경이 강하게 작용하므로 여름철 비닐하우스 내의 기온 상승 및 심한 해충피해가 생장에 영향 주어 유전자형과 환경인자가 상호작용이 크게 발생하였을 것으로 보인다(Day et al., 2002).
수고, 근원경 및 해충피해 회복력에 대하여 GWAS를 실행하였을 때, 수고와 근원경 형질에서는 유의미한 SNP가 확인되지 않았으나, 꼬마버들재주나방 피해 회복력 형질에 대해서는 4번 염색체에서만 유의미한 후보 유전자가 발견되었다(Figure 5, S_Table 1). 해당 유전자는 다른 수종에서도 해충피해 회복력 형질에 대해 관련되어 있다고 보고된 바 없으므로 추후 해당 유전자의 형질 연관성에 대한 검증을 위해 후속 연구가 필요할 것으로 보인다.
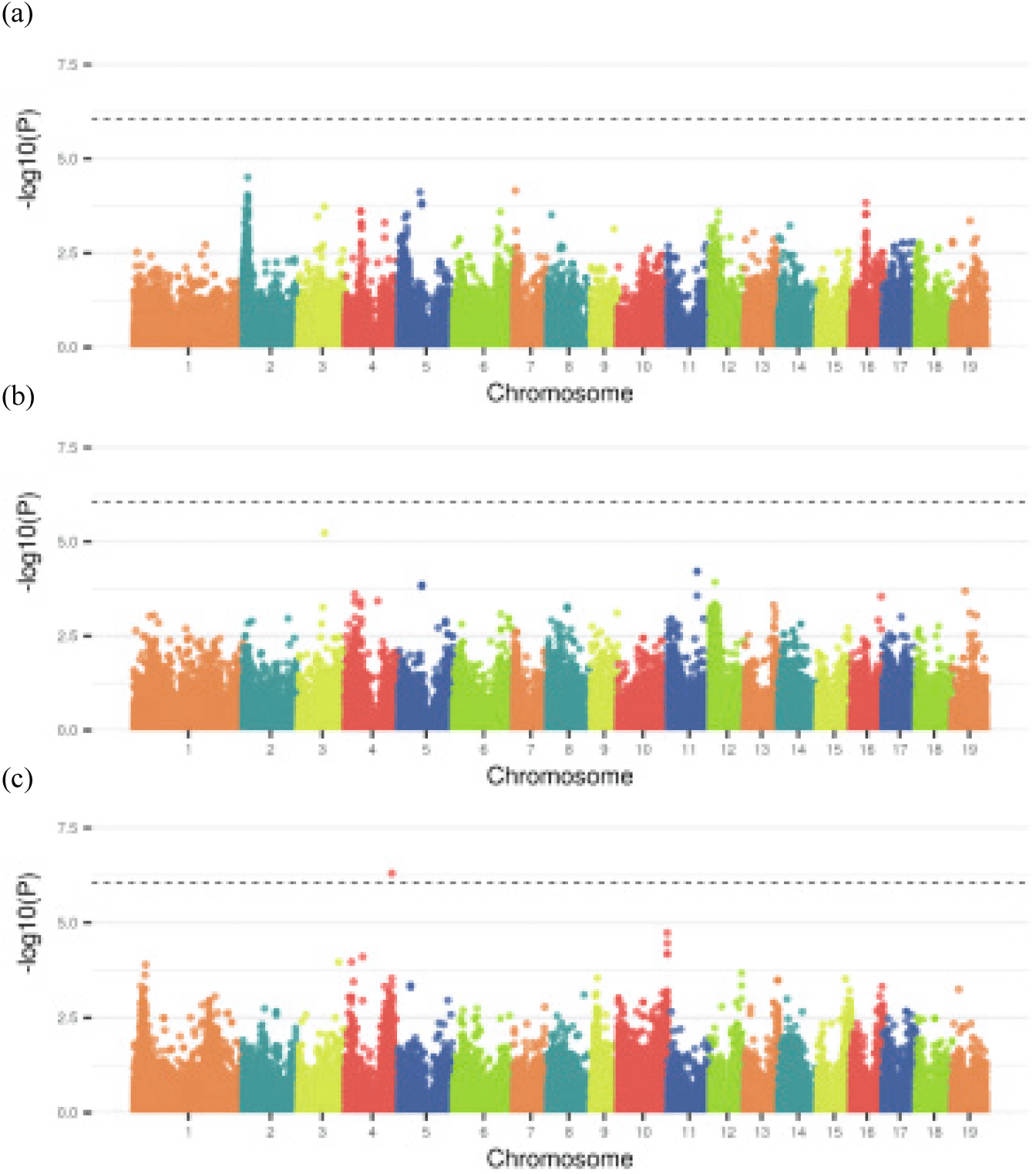
양적형질 유전자좌 탐색과 마찬가지로, p-value에 가장 근접한 값을 가진 유전자 위치를 탐색하여, 이 유전자가 형질과 간접적으로 또는 약하게 연관되어 있다고 추정하였다. 즉, 수고 형질에 대하여 2, 5, 7번 염색체, 근원경 형질에 대하여 3, 5, 11번 염색체 그리고 꼬마버들재주나방 피해 회복력 형질에 대하여는 10번 염색체에서 가장 근접한 p-value를 가지는 유전자를 확인할 수 있었다. 이렇게 GWAS 분석으로 탐색된 유전자는 Table 5와 S_Table 2에 정리하였다.
GWAS 분석을 통하여 꼬마버들재주나방 피해에 따른 해충피해 회복능력 형질에 대해서는 4번 연관군(염색체)에서 연관유전자가 발견되었다. 그러나 GWAS 분석에는 육종집단의 모든 개체가 균일한 정도의 피해를 입었다고 가정하여 분석이 진행되므로 금후 연구에서는 피해 정도에 따른 그룹화 후 분석을 진행하는 것이 필요할 것이다. 특히 근원경 생장은 근계 발달과 관련이 있고, 발달된 근계를 가지고 있는 개체일수록 건조저항성이 높고 척박한 토양에서도 높은 적응력을 보이고 있으므로(Davies and Zhang, 1991; Ericsson et al., 1996) 단순 생장량에 대한 형질로 근원경을 조사하는 것 뿐만 아니라, 환경적응성에 관여하는 형질로서 근원경을 조사할 필요가 있을 것이다. 이러한 형질에 대한 양적형질 유전자위를 탐색하는데 본 연구에서 작성된 사시나무의 고밀도 유전연관지도를 토대로 수행될 수 있을 것으로 보인다. 또한 GWAS로 확인된 해충피해 회복력에 대한 유전자를 확인 및 이용하여 해당 형질에 대한 수형목의 조기선발 및 우수한 사시나무 품종개발 연구 등 사시나무 육종 연구에 도움이 될 수 있을 것으로 기대된다.